EzriCare Eye Drop Recall
In January 2023, the pharmaceutical company EzriPharma announced a voluntary recall of one of its popular eye drop products, EzriCare, due to concerns over potential contamination. The recall was initiated after routine testing detected the...
Nationwide TCPA Class Certified
A Nevada federal court granted class certification on January 10th to customers who received automated text message advertisements after buying tickets from a Las Vegas-based production company. The certified class includes all past, present and...
FDA Warns Patients & Doctors about Transvaginal Mesh
On July 13, 2011, the Food and Drug Administration (FDA) issued an urgent public notice advising patients and their healthcare providers to consider alternatives to transvaginal mesh. The advisory also noted that the FDA will be meeting to discuss...
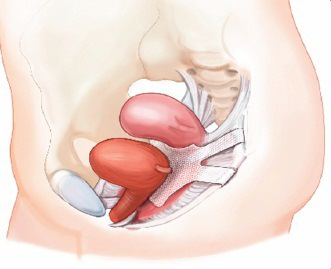
13 Questions FDA Says Patients Should Ask Their Vaginal Mesh Surgeons
On July 13, 2011 the FDA released a Safety Communication regarding the usage of synthetic mesh materials for the repair of pelvic organ prolapse. While the FDA has neither taken the vaginal mesh off the market nor called for a recall, they did...
Public Citizen Calls for Ban on Use of Vaginal Mesh for POP Repair
On August 25, 2011 Public Citizen, a consumer advocacy group representing more than 225,000 members and supporters nationwide, petitioned the Food and Drug Administration (FDA), pursuant to the Medical Device Amendments to the Federal Food, Drug,...
House of Representatives Calls for Hearings on Vaginal Mesh Implants
On January 20, 2012, Representative Henry Waxman of California led House Democrats from the Energy and Commerce Committee in writing a letter to several chairmen, requesting that the Committee hold hearings to review the safety of medical devices...
$40 Million Settlement Against Honda & Acura
A nationwide class action settlement with an estimated value of over $40 million was approved on April 13, 2012 by United States District Judge Katherine S. Hayden of the District of New Jersey, in the case of Alin v. American Honda Motor Co.,...
Federal Judge Certifies CA, NY & FL Class Regarding Honda Tire Wear Defects
On June 12, 2012, a California federal judge struck a blow to Honda, granting class certification to a suit brought by Mr. Mendelsohn alleging the automaker violated warranties and consumer protection laws by knowingly selling Civics that contained...
Honda Civic Premature Tire & Brake Wear
HONDA CIVIC SETTLEMENT We have filed a proposed class action lawsuit against Honda relating to alleged defects in Honda Civic vehicles. Due to the alleged defects with the rear control arms, it is alleged that some Honda Civics are subject to...
Mr. Mendelsohn Named to “New Leaders of the Bar”
In August 2012, Mr. Mendelsohn was named as one of the 50 "New Leaders of the Bar." According to the New Jersey Law Journal, "Mendelsohn is known at his firm as driven, aggressive and mindful that being a successful lawyer means more than just...
